The venous puzzle: from hemodynamics to chronic disease to DVT
Toledo, Ohio, USA; University of Michigan,
Ann Arbor, Michigan, USA
Abstract
The majority of acquired venous disorders are usually classified into two main categories – acute and chronic. Deep vein thrombosis (DVT) exemplifies an acute disease, while morphological and functional abnormalities of the venous system of long duration defines the chronic venous disease (CVD). These definitions, however, resulted from a simplified view on the underlying pathology of these two conditions. Current evidence suggests that the majority of venous thromboses occur in patients already affected by chronic venous disease (CVD). Primary CVD starts early in life, predisposing patients to an acute DVT. At least half of DVTs are subclinical, but they significantly increase the risk of recurrence. Therefore, when a symptomatic DVT occurs, it is more likely to be a stage of chronic venous disease, then an independent event. It is not dissimilar to the relationships between atherosclerosis and acute cardiovascular events, such a stroke or myocardial infarction. The value of recognizing the underlying common pathology of these acute events is in enabling primary and secondary prevention.
Introduction
The majority of acquired venous disorders are usually classified into two main categories – acute and chronic. At first glance, defining these conditions is a simple task. Deep vein thrombosis (DVT), defined as a “formation of thrombi in deep veins…,”1 exemplifies an acute disease. The definition of chronic venous disease (CVD) is “morphological and functional abnormalities of the venous system of long duration.”2 However, a closer look at these definitions reveals that they are not as simple as suggested. For example, it can take a long time for a thrombus to lyse, making the term “acute” imprecise. When the thrombus organizes and transforms into fibrotic tissue, it is no longer appropriate to call it “thrombosis.” At the same time, it may not yet meet the “long duration” criterion of the CVD definition. What is the appropriate category for the condition where the thrombus is rapidly and completely lysed, but damages venous valves and causes vein wall remodeling? These patients can remain asymptomatic for many years or become severely symptomatic within a few months. Does venous disease really need to be “of long duration” to become “chronic”?
What seems to be simply a terminology issue has far-reaching implications. Imprecision in the definitions used in clinical trials leads to inappropriate assignment of patients to the treatment group and erroneous interpretations of the outcomes. Results of observational studies become questionable because misclassification bias within the studies is difficult to assess. Moreover, animal models developed for studying pathological mechanisms of venous disease may lack key relevant features of the disease process. Furthermore, patient management varies significantly depending on the practitioner’s judgment of acuteness or chronicity of the venous condition.
Consensus-based definitions are helpful for current practice and for systematic collection of analyzable data. However, such definitions are frequently based on clinical and phenotypical features of the disease, not on the underlying pathological processes. As our understanding of the disease mechanisms changes, so do consensus based terms. The history of medicine demonstrates many examples of decades- and centuries-long transitions from one set of terminology to another. The field of oncology perhaps best illustrates how an increasing knowledge of cellular and molecular mechanisms of neoplasms results in pathology-based terms to replace the prior clinical terms.
For more than a century, medical students have learned about pathological processes involved in the evolution of a venous thrombus. Unless embolized or lysed, the thrombus undergoes an organization and in a few weeks is replaced by connective tissue. Yet graduates use the terms “chronic thrombosis” and describe the 2-month old process noted in the previous sentence as “thrombus.”
The challenges in connecting the pathological process with clinical manifestations are many. Rethrombosis is one of them. The clinical differentiation between the deterioration of postthrombotic disease and acutely developed rethrombosis is difficult and in many cases impossible. Clinical severity and the timing of clinical manifestations of postthrombotic disease vary from one patient to another, sometimes without an apparent difference in the underlying pathology. Even what appears to be the first episode of acute venous thrombosis in a patient may in fact be a recurrent event, since an estimated 50% of all DVTs are clinically silent.
The last statement deserves some discussion. It migrates within the literature without clear origin or solid confirmation. Despite this, clinical observations consistently support, if not the 50% assertion, the high prevalence of identifiable postthrombotic changes in veins in asymptomatic individuals with no signs of venous disease and no history of DVT.3 It is also a consistent observation that the majority of patients who are accidentally diagnosed with DVT by routine duplex ultrasound surveillance have no symptoms or signs of venous disease.4,5 The annual incidence of acute symptomatic DVT in the United States is estimated to be about 6 in 10 000,6 suggesting that between 100 000 and 200 000 people develop subclinical DVT every year. The more accurate estimate of the DVT incidence in the United States, however, is in the range of 200 000 to 400 000 people per year. In addition, after the first episode of DVT, 7% to 10% of patients develop recurrent thrombosis.7,8 Although the incidence of postthrombotic changes in the veins after acute DVT is unknown, the fact that 40% to 50% of patients with iliofemoral DVT have identifiable postthrombotic changes in their veins after thrombolysis9,10 suggests that it is substantial. Combining the high incidence of asymptomatic DVT and DVT recurrences with the significant frequency of postthrombotic vein changes in these patients (which also significantly increases the risk of recurrent thrombosis11) indicate that many patients who appear to have their first DVT episode have in fact a recurrent event and preexisting subclinical postthrombotic disease. Based on these numbers, at least one-third of all first-time DVT patients have preexisting postthrombotic vein changes. Using the clinical, etiological, anatomical, and pathophysiological (CEAP) classification,12 such patients should be classified as having secondary CVD (Es), even if they have no signs or symptoms (C0, Es, Ad, Po,r). The importance of such a classification is the recognition of an existing pathology. Patients with asymptomatic secondary CVD may remain subclinical for a very long time, but they should not be considered free from venous disease or having a best treatment outcome, especially when enrolled in clinical trials.
Our understanding of the relationship between primary CVD and acute venous thrombosis has also been changing during the last decades. Acute superficial vein thrombosis most frequently occurs in the limbs with varicose veins.13 The common explanation that blood flow disturbances in varicose veins cause the thrombosis neither explains the propagation of the thrombus to and within the saphenous vein nor the cases of thrombosed saphenous veins with no varicose vein involvement. Recent studies demonstrated that endothelium in the area of the valves in superficial veins has unique biological properties protective against thrombosis.14 Due to the vascular remodeling in primary CVD, these areas are affected, increasing the risk of thrombosis.15,16 Varicose veins, a known risk factor for DVT17 are just one of the many manifestations of primary CVD. Pathological studies suggest that primary CVD is a systemic disorder that affects not only superficial veins, but also deep veins and other tissues. Similar pathological changes are present in the wall of varicose veins, in other veins, and even in the skin of distant areas of the body.18,19 Similar to superficial veins, the endothelium of normal deep vein valves has antithrombotic and anti-inflammatory properties.20 Primary CVD alters these properties, and, when combined with venous reflux, leads to activation of prothrombotic and inflammatory pathways.19 Perhaps it should not be surprising that the presence of venous reflux, even without varicose veins, increases the risk of DVT 5-fold.21 Such a strong association suggests that a substantial proportion of patients with DVT have preexisting primary CVD. As in the case of postthrombotic disease, patients with primary CVD may or may not have symptoms and/or clinical signs. Using the CEAP classification, their clinical class can range from C0 to C6, etiology Ep, anatomical component As, Ad, or Asd, and pathophysiology Pr.
Since the time of Trendelenburg and Brodie, studies of primary CVD were predominantly focused on its hemodynamic component. The impact of the failure of venous valves to secure unidirectional flow on venous pressure and blood flow parameters were extensively investigated in basic and clinical research. Accumulated knowledge allowed recent studies to elucidate the biological effects of abnormal venous flow.22 It became clear that normal venous valves create complex hemodynamic phenomena. In vitro and animal models predicted the existence of an isolated stable vortex in the valve pockets of a normal valve, which become hypoxic when the flow lost normal phasicity.23,24 Direct observations confirmed this prediction as well as the active role of the valves in creating and maintaining phasic flow cycle.25,26 These findings substantiate a base for the “valve cusp hypoxia” hypothesis, stating that either hypoxia or reperfusion injury after a prolonged period of hypoxia in the valve pocket plays the key role in the initiation of venous thrombosis.16,27 Abnormal venous valves not only cause venous reflux, but also disrupt the pattern of forward flow.28,29 Computational models based on duplex ultrasound data predicted thrombogenic patterns of blood flow passing the diseased valve, which corresponded to the distribution of platelets in the venous thrombi obtained from autopsy.30 These findings, along with the valve cusp hypoxia hypothesis, explain earlier observations of the sites where venous thrombi originate.31 Interestingly, proposed mechanisms of thrombus initiation do not require mechanical damage of the endothelium and exposure of collagen. All mentioned mechanisms—dysfunctional valves, venous reflux, abnormal flow through the valves, prolonged periods of nonphasic flow, and endothelial dysfunction—are features of primary CVD. Therefore, current basic science evidence not only support biological plausibility of the association between primary CVD and acute DVT, but also suggests that these relationships may be causal.
It is unlikely that primary CVD somehow protects patients with DVT from developing secondary (postthrombotic) CVD; therefore, its prevalence in patients with secondary CVD should be the same or higher than in patients with first-time DVT. Clinical trials and observational studies of postthrombotic syndrome support this statement. An analysis of a large registry of patients with a first episode of DVT showed that close to half of the patients who develop postthrombotic syndrome at 6 months had preexisting symptomatic CVD.32 Of course, the proportion of patients with asymptomatic CVD remains unknown. Unlike secondary CVD that is defined by underlying pathology, the current diagnosis of postthrombotic syndrome is based on a severity score, most frequently using the Villalta scale. Using this score, postthrombotic syndrome has become popular in observational studies because it does not require any objective tests, decreasing cost and simplifying research logistics. However, if signs and symptoms do not reach a certain severity level, a patient is considered “healthy” even if they have occluded iliac veins or a refluxing femoropopliteal venous segment. Thus, the Villalta scale has an exceptionally large misclassification bias,33 making it difficult to differentiate between patients who have true secondary CVD from those whose signs and symptoms after DVT are caused by preexisting primary CVD.
Data from recent randomized trials provide some insight into the true prevalence of preexisting primary CVD in patients with postthrombotic syndrome. The Sox trial (compression StOckings to prevent the post-thrombotic syndrome after symptomatic proXimal deep venous thrombosis) reported 33 patients who developed venous ulcers during the 2-year follow up.34 The ATTRACT trial (Acute venous Thrombosis: Thrombus Removal with Adjunctive Catheterdirected Thrombolysis) reported 29 patients with venous ulcers.35 In both of these trials, the proportion of patients who developed venous ulcers was exactly the same 4%. This is similar to the 3% prevalence of venous ulcers among patients with primary CVD,36 but the timing is very different from studies on the natural history of CVD.37,38 primary CVD starts at a very early age39 and progresses slowly. It takes more than a decade for patients to progress from class C2 to C4, and even longer to develop an ulcer.40 Secondary CVD progresses faster, but still requires more than 5 years to develop venous ulcers.37 In order to observe a 4% ulceration rate in just 2 years, the study patient population has to have an absolute majority of patients with preexisting primary CVD.
It would appear that two pieces of the puzzle are connected. Patients with primary CVD have an increased risk of venous thrombosis. Of those who developed DVT, about half remain subclinical, with a very high risk of symptomatic recurrence. Patients with recurrent DVT are more likely to develop symptomatic secondary CVD. In summary, a large proportion of patients with secondary CVD disease must have preexisting primary CVD, and the natural history of venous disease is full of transitions from chronic conditions to acute events (Figure 1). The CVD itself may be just one disease with two different entry points. It starts in the majority of patients as primary CVD and slowly progresses to different clinical states. Some of these patients undergo one or more episodes of acute thrombosis. In some patients, these episodes significantly increase CVD severity and speed its progression. How frequently and in which patients the DVT episode does not change the CVD dynamics or may even decrease CVD severity remains an open question. Another entry point is acute DVT in patients who do not have preexisting primary CVD. Some of them develop CVD as a sequelae of DVT. Apparently, this happens infrequently. Only 30% of DVT patients develop postthrombotic syndrome,41 and the majority have preexisting primary CVD. The incidence of secondary CVD in patients without primary CVD should not exceed 10%. Future studies will show if this number is even lower.
The “semantic” question of what is acute and what is chronic disease turns out to be a quite complicated subject. Current evidence suggests that the majority of venous thromboses are a stage of chronic venous disease. It is not dissimilar to the relationships between atherosclerosis and acute cardiovascular events, such as stroke or myocardial infarction. The value of recognizing the underlying common pathology of these acute events is in enabling primary and secondary prevention. Separating acute and chronic venous disease creates a barrier for investigating their connection.
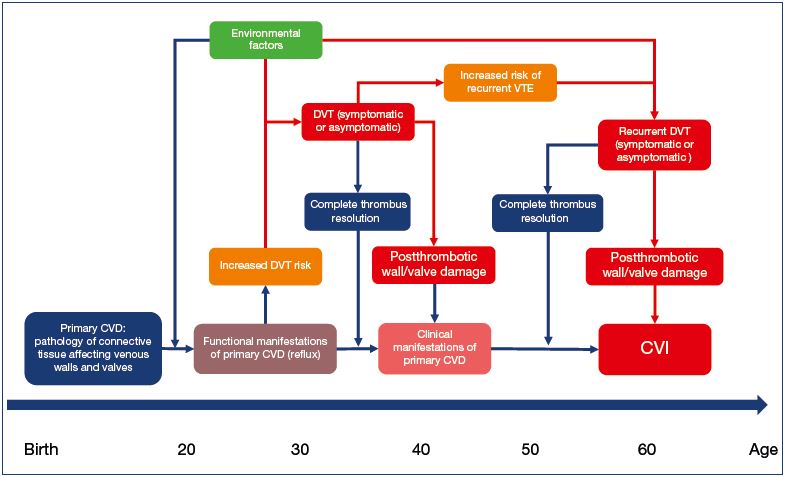
Figure 1. The natural history of chronic venous disease.
Abbreviations: CVD, chronic venous disease; PCVD, primary chronic venous disease; CVI, chronic venous insufficiency;
DVT, deep vein thrombosis.
Almost all studies of mechanisms of venous thrombosis ignore the existence of CVD. Limiting the role of the venous wall and venous valves in the initiation of thrombosis to “endothelial dysfunction” is an inaccurate simplification. Clinical trials routinely ignore the role preexisting CVD plays in the natural history and severity of clinical manifestations. How much of an observed difference in the treatment outcomes of acute DVT should be attributed to the underling CVD remains unknown, making validity of the trials highly debatable. It is time to connect the pieces of the venous disease puzzle, to recognize and investigate in depth the relationships between the “acute” and “chronic” components of this condition.

REFERENCES
1. The Vein Glossary. J Vasc Surg Venous Lymphat Disord. 2018; 6(5): e11-e217.
2. Eklof B, Perrin M, Delis KT, et al. Updated terminology of chronic venous disorders: the VEIN-TERM transatlantic interdisciplinary consensus document. J Vasc Surg. 2009;49(2):498-501.
3. Saarinen J, Kallio T, Lehto M, Hiltunen S, Sisto T. The occurrence of the postthrombotic changes after an acute deep venous thrombosis. A prospective twoyear follow-up study. J Cardiovasc Surg (Torino). 2000;41(3):441-446.
4. Lonner JH, Frank J, McGuire K, Lotke PA. Postthrombotic syndrome after asymptomatic deep vein thrombosis following total knee and hip arthroplasty. Am J Orthop (Belle Mead NJ). 2006;35(10):469-472.
5. van Rij AM, Hill G, Kris J, et al. Prospective study of natural history of deep vein thrombosis: early predictors of poor late outcomes. Ann Vasc Surg. 2013;27(7):924-931.
6. White RH. The epidemiology of venous thromboembolism. Circulation. 2003;107(23 suppl 1):I4-I8.
7. Agnelli G, Buller HR, Cohen A, et al. Apixaban for extended treatment of venous thromboembolism. N Engl J Med. 2013;368(8):699-708.
8. Bauersachs R, Berkowitz SD, Brenner B, et al; EINSTEIN Investigators. Oral rivaroxaban for symptomatic venous thromboembolism. N Engl J Med. 2010;363(26):2499-2510.
9. Enden T, Haig Y, Klow ND, et al. Longterm outcome after additional catheterdirected thrombolysis versus standard treatment for acute iliofemoral deep vein thrombosis (the CaVenT study): a randomised controlled trial. Lancet. 2012;379(9810):31-38.
10. Lu Y, Chen L, Chen J, Tang T. Catheterdirected thrombolysis versus standard anticoagulation for acute lower extremity deep vein thrombosis: a meta-analysis of clinical trials. Clin Appl Thromb Hemost. 2018;24(7):1134-1143.
11. Aziz F, Comerota AJ. Quantity of residual thrombus after successful catheterdirected thrombolysis for iliofemoral deep venous thrombosis correlates with recurrence. Eur J Vasc Endovasc Surg. 2012;44(2):210-213.
12. Eklof B, Rutherford RB, Bergan JJ, et al. Revision of the CEAP classification for chronic venous disorders: consensus statement. J Vasc Surg. 2004;40(6):1248-1252.
13. Lurie F. Acute superficial thrombophlebitis of the great saphenous vein. In: Upchurch GR, Henke PK, eds. Clinical Scenarios in Vascular Surgery. 2nd ed. Philadelphia, PA: Lippincott Williams & Wilkins; 2005:522-524.
14. Brooks EG, Trotman W, Wadsworth MP, et al. Valves of the deep venous system: an overlooked risk factor. Blood. 2009;114(6):1276-1279.
15. Takase S, Pascarella L, Lerond L, et al. Venous hypertension, inflammation and valve remodeling. Eur J Vasc Endovasc Surg. 2004;28(5):484-493.
16. Esmon CT. Basic mechanisms and pathogenesis of venous thrombosis. Blood Rev. 2009;23(5):225-229.
17. Chang SL, Huang YL, Lee MC, et al. Association of varicose veins with incident venous thromboembolism and peripheral artery disease. JAMA. 2018;319(8):807-817.
18. Sansilvestri-Morel PR, Badier-Commander A, Kern C, et al. Imbalance in the synthesis of collagen type I and collagen type III in smooth muscle cells derived from human varicose veins. J Vasc Res. 2001;38(6):560-568.
19. Kockx MM, Knaapen MW, Bortier HE, et al. Vascular remodeling in varicose veins. Angiology. 1998;49(11):871-7.
20. Shaydakov M. 2016 BSN Jobst Research Grant – Final Report. In: 30th American Venous Forum Annual Meeting. 2018. Tucson, AZ.
21. Shaydakov ME., Comerota AJ, Lurie F. Primary venous insufficiency increases risk of deep vein thrombosis. J Vasc Surg Venous Lymphat Disord. 2016;4(2):161- 166.
22. Bergan JJ, Schmid-Schonbein GW, Smith PD, et al. Chronic venous disease. N Engl J Med. 2006;355(5):488-498.
23. Karino T, Motomiya M. Flow through a venous valve and its implication for thrombus formation. Thromb Res. 1984;36(3):245-257.
24. Hamer JD, Malone PC, Silver IA. The PO2 in venous valve pockets: its possible bearing on thrombogenesis. Br J Surg. 1981;68(3):166-170.
25. Lurie F, Kistner RL, Eklof B. The mechanism of venous valve closure in normal physiologic conditions. J Vasc Surg. 2002;35(4):713-717.
26. Lurie F, Kistner RL, Eklof B, Kessler D. Mechanism of venous valve closure and role of the valve in circulation: a new concept. J Vasc Surg. 2003;38(5):955- 961.
27. Malone P Colm, Agutter PS. The Aetiology of Deep Venous Thrombosis: a Critical, Historical and Epistemological Survey. Dordrecht, Netherlands: Springer; 2008.
28. Lurie F, Kistner RL. On the existence of helical flow in veins of the lower extremities. J Vasc Surg Venous Lymphat Disord. 2013;1(2):134-138.
29. Chen HY, Diaz JA, Lurie F, Chambers SD, Kassab GS. Hemodynamics of venous valve pairing and implications on helical flow. J Vasc Surg Venous Lymphat Disord. 2018;6(4):517-522 e1.
30. Bajd F, Vidmar J, Fabian A, et al. Impact of altered venous hemodynamic conditions on the formation of platelet layers in thromboemboli. Thromb Res. 2012;129(2):158-163.
31. Sevitt S. The structure and growth of valve-pocket thrombi in femoral veins. J Clin Pathol. 1974;27(7):517-528.
32. Galanaud JP, Holcroft CA, Rodger MA, et al. Comparison of the Villalta post-thrombotic syndrome score in the ipsilateral vs. contralateral leg after a first unprovoked deep vein thrombosis. J Thromb Haemost. 2012;10(6):1036- 1042.
33. Trinh FP, Fish D, Kasper J, Lurie F. Use of Villalta score for defining postthrombotic disease may lead to falsepositive diagnosis in 42% of patients with primary chronic venous disease. J Vasc Surg Venous Lymphat Disord. 2018;6(2):291.
34. Kahn SR, Shapiro S, Wells PS, et al; SOX Trial Investigators. Compression stockings to prevent post-thrombotic syndrome: a randomised placebo-controlled trial. Lancet. 2014;383(9920):880-888.
35. Vedantham S, Goldhaber SZ, Julian JA, et al; ATTRACT Trial Investigators. Pharmacomechanical catheter-directed thrombolysis for deep-vein thrombosis. N Engl J Med. 2017;377(23):2240- 2252.
36. Rabe E, Bromen K, Schuldt K, et al. Bonner Venenstudie der Deutschen Gesellschaft für Phlebologie – Epidemiologische Untersuchung zur Frage der Häufigkeit und Ausprägung von Chronischen Venenkrankheiten in der städtischen und ländlichen Wohnbevölkerung. Phlebologie. 2003;32:1-14.
37. Lurie F, Makarova NP, Hmelniker SM. Development of postthrombotic syndrome after acute unilateral iliofemoral thrombosis: clinical dynamics and hemodynamic changes. Vasc Surg. 1999;33:5-13.
38. Lurie, FM, Makarova NP. Clinical dynamics of varicose disease in patients with high degree of venous reflux during conservative treatment and after surgery: 7-year follow-up. Int J Angiol. 1998;7(3):234-237.
39. Schultz-Ehrenburg U, Weindorf N, Matthes U, Hirche H. An epidemiologic study of the pathogenesis of varices. The Bochum study I-IIIn [article in French]. Phlebologie. 1992;45(4):497-500.
40. Lee AJ, Robertson LA, Boghossian SM, et al. Progression of varicose veins and chronic venous insufficiency in the general population in the Edinburgh Vein Study. J Vasc Surg Venous Lymphat Disord. 2015;3(1):18-26.
41. Prandoni P, Lensing AW, Cogo A, et al. The long-term clinical course of acute deep venous thrombosis. Ann Intern Med. 1996;125(1): 1-7.